肯德尔τ检验#
肯德尔τ是衡量两个排名之间对应关系的一种度量。
考虑来自[1]的以下数据,该研究探讨了不健康人肝脏中游离脯氨酸(一种氨基酸)与总胶原蛋白(一种常存在于结缔组织中的蛋白质)之间的关系。
下面的x和y数组记录了这两种化合物的测量值。观测值是配对的:每个游离脯氨酸测量值都与相同索引处的总胶原蛋白测量值来自同一肝脏。
import numpy as np
# total collagen (mg/g dry weight of liver)
x = np.array([7.1, 7.1, 7.2, 8.3, 9.4, 10.5, 11.4])
# free proline (μ mole/g dry weight of liver)
y = np.array([2.8, 2.9, 2.8, 2.6, 3.5, 4.6, 5.0])
这些数据在[2]中使用斯皮尔曼秩相关系数进行了分析,该统计量与肯德尔τ相似,因为它也对样本之间的顺序相关性敏感。让我们使用Kendall's tau进行类似的研究。
from scipy import stats
res = stats.kendalltau(x, y)
res.statistic
np.float64(0.5499999999999999)
对于具有强正序数相关性的样本,该统计量的值倾向于高(接近 1);对于具有强负序数相关性的样本,该值倾向于低(接近 -1);对于序数相关性较弱的样本,其值大小接近零。
该检验通过将统计量的观测值与零分布(在总胶原蛋白和游离脯氨酸测量值独立的零假设下导出的统计量值分布)进行比较来执行。
对于此检验,无系大样本的零分布近似于正态分布,其方差为(2*(2*n + 5))/(9*n*(n - 1)),其中n = len(x)。
import matplotlib.pyplot as plt
n = len(x) # len(x) == len(y)
var = (2*(2*n + 5))/(9*n*(n - 1))
dist = stats.norm(scale=np.sqrt(var))
z_vals = np.linspace(-1.25, 1.25, 100)
pdf = dist.pdf(z_vals)
fig, ax = plt.subplots(figsize=(8, 5))
def plot(ax): # we'll reuse this
ax.plot(z_vals, pdf)
ax.set_title("Kendall Tau Test Null Distribution")
ax.set_xlabel("statistic")
ax.set_ylabel("probability density")
plot(ax)
plt.show()
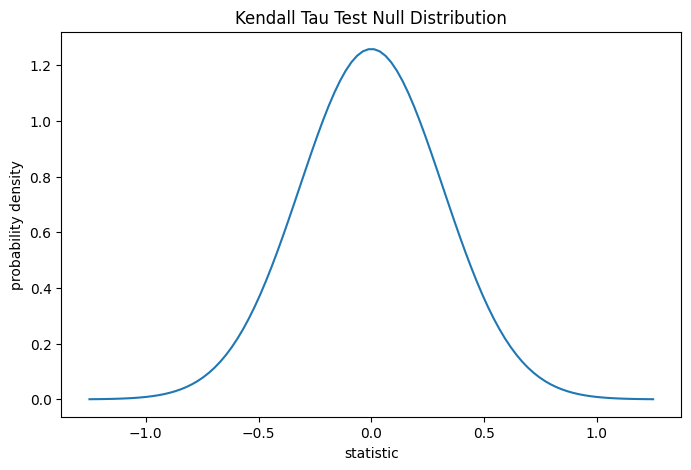
这种比较通过 p 值量化:p 值是零分布中与统计量观测值一样极端或更极端的值的比例。在双边检验中,如果统计量为正,则零分布中大于转换后统计量的元素和小于观测统计量负值的元素都被认为是“更极端”的。
fig, ax = plt.subplots(figsize=(8, 5))
plot(ax)
pvalue = dist.cdf(-res.statistic) + dist.sf(res.statistic)
annotation = (f'p-value={pvalue:.4f}\n(shaded area)')
props = dict(facecolor='black', width=1, headwidth=5, headlength=8)
_ = ax.annotate(annotation, (0.65, 0.15), (0.8, 0.3), arrowprops=props)
i = z_vals >= res.statistic
ax.fill_between(z_vals[i], y1=0, y2=pdf[i], color='C0')
i = z_vals <= -res.statistic
ax.fill_between(z_vals[i], y1=0, y2=pdf[i], color='C0')
ax.set_xlim(-1.25, 1.25)
ax.set_ylim(0, 0.5)
plt.show()
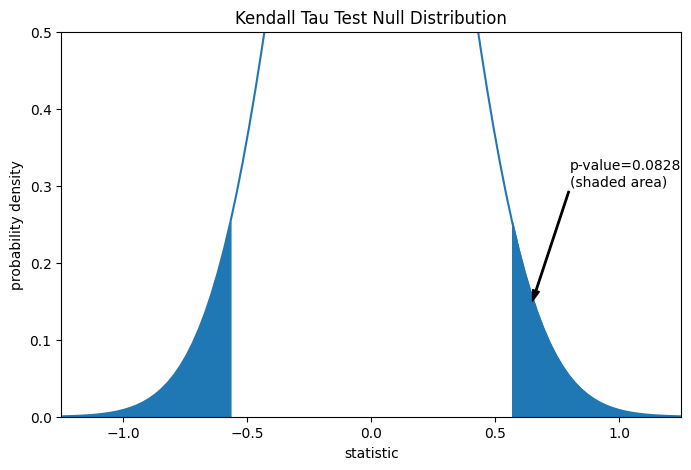
res.pvalue
np.float64(0.09108705741631495)
请注意,曲线的阴影区域与scipy.stats.kendalltau返回的 p 值之间存在轻微差异。这是因为我们的数据存在系,并且我们忽略了scipy.stats.kendalltau执行的对零分布方差的系校正。对于无系样本,我们图的阴影区域和scipy.stats.kendalltau返回的 p 值将完全匹配。
如果 p 值“很小”——也就是说,从独立分布中抽样数据产生如此极端统计量值的概率很低——这可以作为反对零假设的证据,转而支持备择假设:总胶原蛋白和游离脯氨酸的分布不是独立的。请注意
反之则不成立;也就是说,此检验不能用于为零假设提供证据。
将被视为“小”值的阈值应在数据分析之前做出选择[3],并考虑假阳性(错误地拒绝零假设)和假阴性(未能拒绝错误的零假设)的风险。
小的 p 值不是大效应的证据;相反,它们只能提供“显著”效应的证据,这意味着它们在零假设下不太可能发生。
对于中等大小的无系样本,scipy.stats.kendalltau可以精确计算 p 值。然而,在存在系的情况下,scipy.stats.kendalltau会采用渐近近似。尽管如此,我们可以使用置换检验精确计算零分布:在总胶原蛋白和游离脯氨酸独立的零假设下,每个游离脯氨酸测量值与任何总胶原蛋白测量值一起被观测到的可能性是均等的。因此,我们可以通过计算x和y之间每种可能的元素配对下的统计量来形成一个精确的零分布。
def statistic(x): # explore all possible pairings by permuting `x`
return stats.kendalltau(x, y).statistic # ignore pvalue
ref = stats.permutation_test((x,), statistic,
permutation_type='pairings')
fig, ax = plt.subplots(figsize=(8, 5))
plot(ax)
bins = np.linspace(-1.25, 1.25, 25)
ax.hist(ref.null_distribution, bins=bins, density=True)
ax.legend(['asymptotic approximation\n(many observations)',
'exact null distribution'])
plot(ax)
plt.show()
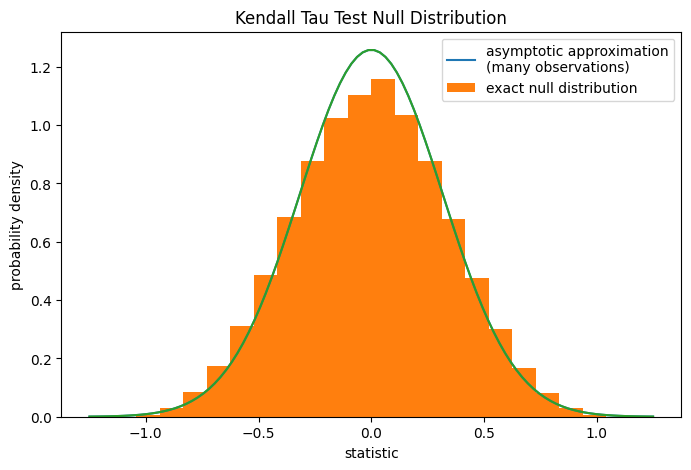
ref.pvalue
np.float64(0.12222222222222222)
请注意,此处计算的精确 p 值与上述scipy.stats.kendalltau返回的近似值之间存在显著差异。对于带系的小样本,请考虑执行置换检验以获得更准确的结果。